How does a molecule break apart in an intense laser field and what sequential processes take place how quickly? Physicists at the Heidelberg MPI for Nuclear Physics (MPIK) have investigated this question in collaboration with a research group from Ottawa in Canada with a new method – studying the example of the hydrogen molecule H2. To do this, they use extremely short laser flashes on the order of femtoseconds (fs, a millionth of a billionth of a second). Such laser pulses also play a key role in controlling molecular reactions, as they directly influence the dynamics of the electrons responsible for chemical bonding.
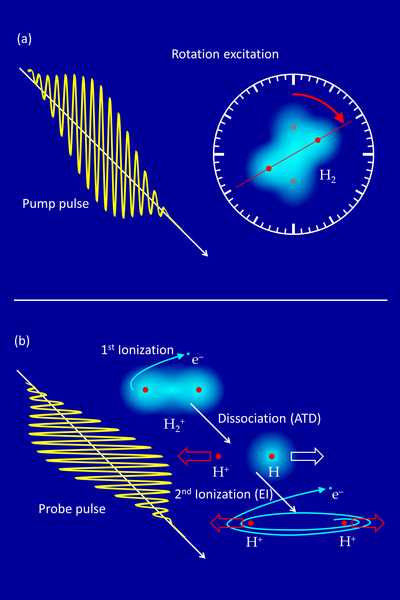
If a hydrogen molecule (H2) is exposed to a strong infrared laser flash (800 nm wavelength) of a few 1014 W/cm2 intensity, the electric field of the laser first rips off one of the two electrons. More than 10 photons are absorbed at the same time in this ionization process. The remaining molecular ion H2+ with only one electron is no longer in equilibrium and becomes stretched due to the repulsion of the two protons. By absorbing further photons, it can break up into a proton (H+) and a neutral hydrogen atom (H). This reaction is called “above threshold dissociation” (ATD). If the molecular ion is stretched further to a nuclear distance of a few atomic radii, the remaining electron can absorb energy resonantly by the laser field, as in a small antenna, and is eventually also released. This mechanism is called "enhanced ionization" (EI). It leads to the “Coulomb explosion” of the two repelling protons.
The researchers investigate these processes at the MPIK's laser laboratory using a reaction microscope, which allows for the detection of all charged fragments (protons, electrons) after the break-up of the molecule. The femtosecond laser pulses are focused onto a thin supersonic beam of hydrogen molecules in order to achieve the desired intensity. Protons from the ATD and EI processes can be distinguished via their kinetic energy.
Obviously, EI takes a little more time than ATD – but how much and can this be measured? Here, a problem arises since the laser pulse has to last long enough (approx. 25 fs) to start these processes, but has to be short enough to extract precise time information (a few fs). Since this cannot be realized in one single laser pulse, the researchers used the following trick: In principle, every molecule possesses a kind of “internal clock” since it can be stimulated to rotate.
A first (slightly weaker) pump pulse excites the molecular rotation (Fig. 1a), followed with a variable time delay by a second (slightly stronger) probe pulse triggering the fragmentation (ATD or EI) (Fig. 1b). Both processes are sensitive to the orientation of the molecular axis relative to the plane in which the electric field oscillates – they are most likely for parallel orientation. The two laser pulses are linearly polarized perpendicular to each other in order to sort out fragmentation events from the first pulse.
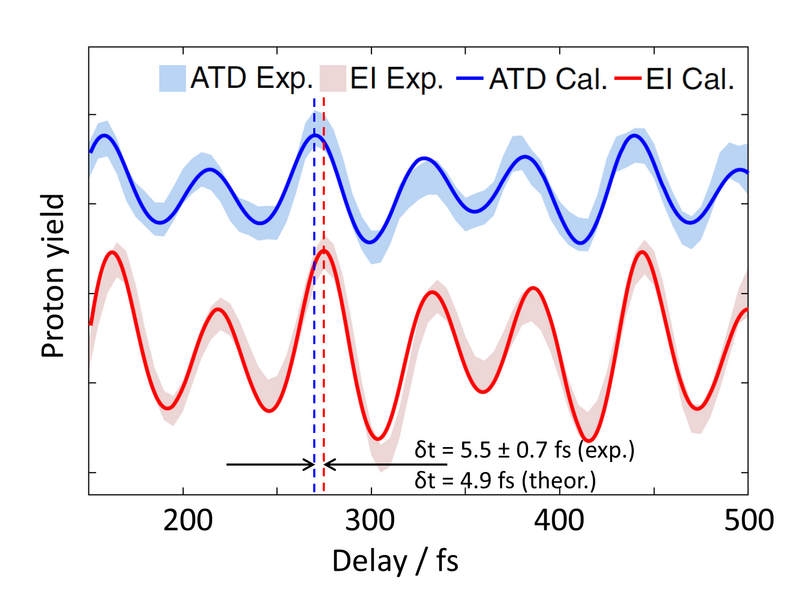
The experimental yield of ATD and EI events shows an almost regular up and down, corresponding to the rotation of the molecule (Fig. 2). In a closer analysis, however, a slight delay of approx. 5.5 fs is observed for EI compared to ATD. This is the typical time that the molecular ion needs to stretch until the electron couples resonantly to the laser field. Using theoretical model calculations, further details can be extracted and the experimental results are very well reproduced. The experiment was also carried out with the heavier isotope deuterium (D2). Here, the delay is found to be approx. 6.5 fs. This is slightly less than the value expected based on the mass ratio (factor √2). The reason is the slower motion of D2+, which reaches the EI region after approx. 20 fs – for this, there is hardly enough time during a 25 fs laser pulse.
The method of a “rotation clock” can – in principle – be applied to similar multi-step reactions in other molecules and thus possibly even forms the basis for a general approach to the control of molecular dynamics.
mpik
Original publication:
Clocking enhanced ionization of hydrogen molecules with rotational wavepackets
Y. Mi, P. Peng, N. Camus, X. Sun, P. Fross, D. Martinez, Z. Dube, P. B. Corkum, D. M. Villeneuve, A. Staudte, R. Moshammer and T. Pfeifer
Phys. Rev. Lett. 125, 173201 (2020), DOI: 10.1103/PhysRevLett.125.173201
Weblinks:
Division "Quantum Dynamics and Control" at MPIK